The Acetaminophen Molecule-Tylenol
Ball and Stick Model for Acetaminophen Molecule
To View the Acetaminophen Molecule in 3D --->>in 3D with Jmol
Chemical and Physical Properties of Tylenol Molecule-Acetaminophen
Paracetamol or Acetaminophen
Paracetamol or acetaminophen (USAN), is the active metabolite of phenacetin, a so-called coal tar analgesic. Unlike phenacetin, paracetamol has not been shown to be carcinogenic in any way. It has analgesic and antipyretic properties, but, unlike aspirin, it is not a very effective anti-inflammatory agent. It is well tolerated, lacks many of the side-effects of aspirin, and is available over-the-counter, so it is commonly used for the relief of fever, headaches, and other minor aches and pains. Paracetamol is also useful in the management of more severe pain, where it allows lower dosages of additional non-steroidal anti-inflammatory drugs (NSAIDs) or opioid analgesics to be used, thereby minimizing overall side-effects. It is a major ingredient in numerous cold and flu medications, including Tylenol and Panadol, among others. It is considered safe for human use at recommended doses; however, acute overdose can cause fatal liver damage often heightened with use of alcohol, and the number of accidental self-poisonings and suicides has grown in recent years.
The words acetaminophen and paracetamol come from the chemical names for the compound: para-acetylaminophenol and para-acetylaminophenol. (The brand name Tylenol also derives from this name: para-acetylaminophenol.) In some contexts, it is shortened to APAP, for N-acetyl-para-aminophenol.
History
In ancient and medieval times, known antipyretic agents were compounds contained in white willow bark (a family of chemicals known as salicins, which led to the development of aspirin), and compounds contained in cinchona bark.[1] Cinchona bark was also used to create the anti-malaria drug quinine. Quinine itself also has antipyretic effects. Efforts to refine and isolate salicin and salicylic acid took place throughout the middle- and late-19th century, and was accomplished by Bayer chemist Felix Hoffmann (this was also done by French chemist Charles Frederic Gerhardt 40 years earlier, but he abandoned the work after deciding it was impractical).[2]
When the cinchona tree became scarce in the 1880s, people began to look for alternatives. Two alternative antipyretic agents were developed in the 1880s: acetanilide in 1886 and phenacetin in 1887. Harmon Northrop Morse first synthesized paracetamol via the reduction of p-nitrophenol with tin in glacial acetic acid in 1878; however, paracetamol was not used in medical treatment for another 15 years. In 1893, paracetamol was discovered in the urine of individuals that had taken phenacetin, and was concentrated into a white, crystalline compound with a bitter taste. In 1899, paracetamol was found to be a metabolite of acetanilide. This discovery was largely ignored at the time.
In 1946, the Institute for the Study of Analgesic and Sedative Drugs awarded a grant to the New York City Department of Health to study the problems associated with analgesic agents. Bernard Brodie and Julius Axelrod were assigned to investigate why non-aspirin agents were associated with the development of methemoglobinemia, a condition that decreases the oxygen-carrying capacity of blood and is potentially lethal. In 1948, Brodie and Axelrod linked the use of acetanilide with methemoglobinemia and determined that the analgesic effect of acetanilide was due to its active metabolite paracetamol. They advocated the use of paracetamol, since it did not have the toxic effects of acetanilide.[3]
The product was first sold in 1955 by McNeil Laboratories as a pain and fever reliever for children, under the brand name Tylenol Children's Elixir.[4]
In 1956, 500 mg tablets of paracetamol went on sale in the United Kingdom under the trade name Panadol, produced by Frederick Stearns & Co, a subsidiary of Sterling Drug Inc. Panadol was originally available only by prescription, for the relief of pain and fever, and was advertised as being "gentle to the stomach," since other analgesic agents of the time contained aspirin, a known stomach irritant. In June 1958, a children's formulation, Panadol Elixir, was released.
In 1963, paracetamol was added to the British Pharmacopoeia, and has gained popularity since then as an analgesic agent with few side-effects and little interaction with other pharmaceutical agents.
The U.S. patent on paracetamol has long expired, and generic versions of the drug are widely available under the Drug Price Competition and Patent Term Restoration Act of 1984, although certain Tylenol preparations were protected until 2007. U.S. patent 6,126,967 filed September 3, 1998 was granted for "Extended release acetaminophen particles."
Chemical Properties
Structure and reactivity
Paracetamol consists of a benzene ring core, substituted by one hydroxyl group and the nitrogen atom of an amide group in the para (1,4) pattern. The amide group is acetamide (ethanamide). It is an extensively conjugated system, as the lone pair on the hydroxyl oxygen, the benzene pi cloud, the nitrogen lone pair, the p orbital on the carbonyl carbon, and the lone pair on the carbonyl oxygen are all conjugated. The presence of two activating groups also make the benzene ring highly reactive toward electrophilic aromatic substitution. As the substituents are ortho,para-directing and para with respect to each other, all positions on the ring are more or less equally activated. The conjugation also greatly reduces the basicity of the oxygens and the nitrogen, while making the hydroxyl acidic through delocalisation of charge developed on the phenoxide anion.
Synthesis
From the starting material phenol, paracetamol can be made in the following manner:
Phenol is nitrated using sulfuric acid and sodium nitrate (as phenol is highly activated, its nitration requires very mild conditions compared to the oleum-fuming nitric acid mixture required to nitrate benzene).
The para isomer is separated from the ortho isomer by fractional distillation (there will be little of meta, as OH is o-p directing).
The 4-nitrophenol is reduced to 4-aminophenol using a reducing agent such as sodium borohydride in basic medium.
The 4-nitrophenol is reduced to 4-aminophenol using a reducing agent such as sodium borohydride in basic medium
4-aminophenol is reacted with acetic anhydride to give paracetamol.
Notice that the synthesis of paracetamol lacks one very significant difficulty inherent in almost all drug syntheses: Lack of stereocenters means there is no need to design a stereo-selective synthesis. More efficient, industrial syntheses are also available.
Available forms
Panadol, which is marketed in Europe, Africa, Asia, Central America, and Australia, is the most widely available brand, sold in over 80 countries. In North America, paracetamol is sold in generic form (usually labeled as acetaminophen) or under a number of trade names, for instance, Tylenol (McNeil-PPC, Inc), Anacin-3, Tempra, and Datril.
In some formulations, paracetamol is combined with the opioid codeine, sometimes referred to as co-codamol (BAN). In the United States and Canada, this is marketed under the name of Tylenol #1/2/3/4, which contain approximately 1/8 grain, approximately 1/4 grain, approximately 1/2 grain, and approximately 1 grain of codeine, respectively. A US grain is 64.78971 milligrams - this is usually rounded in manufacture down to a multiple of 5 mg (so that a #3 contains 30 mg, and a #4 contains 60 mg, while a #1 may be 8 mg or 10 mg depending on manufacturer. In the U.S., this combination is available only by prescription, while the lowest-strength preparation is over-the-counter in Canada, and, in other countries, other strengths may be available over the counter. There are generics as well. In the UK and in many other countries, this combination is marketed under the names of Tylex CD and Panadeine. Other names include Captin, Disprol, Dymadon, Fensum, Hedex, Mexalen, Nofedol, Paralen, Pediapirin, Perfalgan, and Solpadeine. Paracetamol is also combined with other opioids such as dihydrocodeine, referred to as co-dydramol (BAN), oxycodone or hydrocodone, marketed in the U.S. as Percocet and Vicodin, respectively. Another very commonly used analgesic combination includes paracetamol in combination with propoxyphene napsylate, sold under the brand name Darvocet. A combination of paracetamol, codeine, and the calmative doxylamine succinate is marketed as Syndol or Mersyndol. Paracetamol is commonly used in multi-ingredient preparations for migraine headache, typically including butalbital and paracetamol with or without caffeine, and sometimes containing codeine.
It is commonly administered in tablet, liquid suspension, suppository, intravenous, or intramuscular form. The common adult dose is 500 mg to 1000 mg. The recommended maximum daily dose, for adults, is 4 grams. In recommended doses, paracetamol is safe for children and infants, as well as for adults.
Brand Names
Aceta, Actimin, Anacin-3, Apacet, Aspirin Free Anacin, Atasol, Banesin, Dapa, Datril Extra-Strength, Feverall, Genapap, Genebs, Liquiprin, Neopap, Oraphen-PD, Panadol, Phenaphen, Redutemp, Snaplets-FR, Suppap, Tapanol, Tylenol, Valorin
Mechanism of action
The mechanism by which paracetamol reduces fever and pain is still a source of debate[citation needed]. The reason for this confusion has largely been due to the fact that paracetamol reduces the production of prostaglandins, pro-inflammatory chemicals the production of which is also inhibited by aspirin, but, unlike aspirin, paracetamol does not have much anti-inflammatory action. Likewise, whereas aspirin inhibits the production of the pro-clotting chemicals thromboxanes, paracetamol does not. Aspirin is known to inhibit the cyclooxygenase (COX) family of enzymes, and, because of paracetamol's partial similarity of aspirin's action, much research has focused on whether paracetamol also inhibits COX. It is now clear, however, that paracetamol acts via (at least) two pathways.[6][7][8][9]
The COX family of enzymes are responsible for the metabolism of arachidonic acid to prostaglandin H2, an unstable molecule, which is, in turn, converted to numerous other pro-inflammatory compounds. Classical anti-inflammatories, such as the NSAIDs, block this step. The activity of the COX enzyme relies on its being in the oxidized form to be specific, tyrosine 385 must be oxidized to a radical.[10][11] It has been shown that paracetamol reduces the oxidized form of the COX enzyme, preventing it from forming pro-inflammatory chemicals.[7][12]
Further research has shown that paracetamol also modulates the endogenous cannabinoid system.[13] Paracetamol is metabolized to AM404, a compound with several actions; most important, it inhibits the uptake of the endogenous cannabinoid/vanilloid anandamide by neurons. Anandamide uptake would result in the activation of the main pain receptor (nociceptor) of the body, the TRPV1 (older name: vanilloid receptor). Furthermore, AM404 inhibits sodium channels such as anesthetics, lidocaine and procaine.[14] Either of these actions by themselves has been shown to reduce pain, and are a possible mechanism for paracetamol, though it has been demonstrated that, after blocking cannabinoid receptors and hence making any action of cannabinoid reuptake irrelevant, paracetamol no longer has any analgesic effect, suggesting its pain-relieving action is indeed mediated by the endogenous cannabinoid system.[15]
A theory that held some sway, but has now largely been discarded, is that paracetamol inhibits the COX-3 isoform of the cyclooxygenase family of enzymes.[6][16] This enzyme, when expressed in dogs, shares a strong similarity to the other COX enzymes, produces pro-inflammatory chemicals, and is selectively inhibited by paracetamol. However, in humans and mice, the COX-3 enzyme is without inflammatory action, and is not modulated by paracetamol.[6]
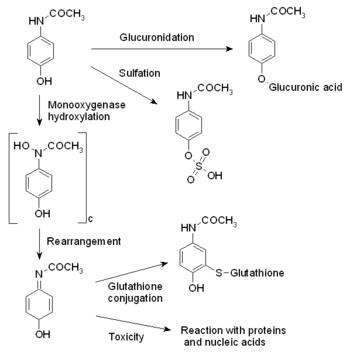
The reactions involved in paracetamol metabolism.
Paracetamol is metabolised primarily in the liver, where its major metabolites include inactive sulfate and glucuronide conjugates, which are excreted by the kidneys. Only a small, yet significant amount is metabolised via the hepatic cytochrome P450 enzyme system (its CYP2E1 and CYP1A2 isoenzymes), which is responsible for the toxic effects of paracetamol due to a minor alkylating metabolite (N-acetyl-p-benzo-quinone imine, abbreviated as NAPQI).[17] There is a great deal of polymorphism in the P450 gene, and genetic polymorphisms in CYP2D6 have been studied extensively. The population can be divided into "extensive," "ultrarapid," and "poor metabolizers" depending on their levels of CYP2D6 expression. CYP2D6 may also contribute to the formation of NAPQI, albeit to a lesser extent than other P450 isozymes, and its activity may contribute to paracetamol toxicity, in particular, in extensive and ultrarapid metabolizers and when paracetamol is taken at very large doses.[18]
The metabolism of paracetamol is an excellent example of toxication, because the metabolite NAPQI is primarily responsible for toxicity rather than paracetamol itself.
Paracetamol overdose results in more calls to poison control centers in the US than overdose of any other pharmacological substance, accounting for more than 100,000 calls, as well as 56,000 emergency room visits, 2,600 hospitalizations, and 458 deaths due to acute liver failure per year.[19] A recent study of cases of acute liver failure between November 2000 and October 2004 by the Centers for Disease Control and Prevention (US) found that paracetamol was the cause of 41% of all cases in adults, and 25% of cases in children.[20]
At usual doses, the toxic metabolite NAPQI is quickly detoxified by combining irreversibly with the sulfhydryl groups of glutathione or administration of a sulfhydryl compound such as N-acetylcysteine, to produce a non-toxic conjugate that is eventually excreted by the kidneys.[17] Also, methionine has been recommended in some cases,[21] although recent studies show that N-acetylcysteine is a more effective antidote to paracetamol overdose.[22]
Comparison with NSAIDs
Paracetamol, unlike other common analgesics such as aspirin and ibuprofen, has relatively little anti-inflammatory activity, and so it is not considered to be a non-steroidal anti-inflammatory drug (NSAID).
Efficacy
Regarding comparative efficacy, studies show conflicting results when compared to NSAIDs. A randomized controlled trial of chronic pain from osteoarthritis in adults found similar benefit from acetaminophen and ibuprofen.[23] However, a randomized controlled trial of acute musculoskeletal pain in children found that the standard OTC dose of ibuprofen (400 mg) gives greater relief of pain than the standard dose of paracetamol (1000 mg).[24]
Adverse effects
In recommended doses, paracetamol does not irritate the lining of the stomach, affect blood coagulation as much as NSAIDs, or affect function of the kidneys. However, some studies have shown that high dose-usage (greater than 2000 mg per day) does increase the risk of upper gastrointestinal complications.[25] Paracetamol is safe in pregnancy, and does not affect the closure of the fetal ductus arteriosus as NSAIDs can. Unlike aspirin, it is safe in children, as paracetamol is not associated with a risk of Reye's syndrome in children with viral illnesses.Like NSAIDs and unlike opioid analgesics, paracetamol has not been found to cause euphoria or alter mood in any way. Paracetamol and NSAIDs have the benefit of bearing a low risk of addiction, dependence, tolerance, and withdrawal, but, unlike opioid medications, may damage the liver; however, this is, in general, taken into account when compared to the danger of addiction. Paracetamol, particularly in combination with weak opioids, is more likely than NSAIDs to cause rebound headache (medication overuse headache), although less of a risk than ergotamine or triptans used for migraines.[26]
Toxicity
Paracetamol is contained in many preparations (both over-the-counter and prescription-only medications). In some animals, for example, cats, small doses are toxic. Because of the wide availability of paracetamol, there is a large potential for overdose and toxicity.[27] Without timely treatment, overdose can lead to liver failure and death within days; paracetamol toxicity is, by far, the most common cause of acute liver failure in both the United States and the United Kingdom.[28][29] It is sometimes used in suicide attempts by those unaware of the prolonged timecourse and high morbidity (likelihood of significant illness) associated with paracetamol-induced toxicity in survivors.
In the UK, sales of over-the-counter paracetamol are restricted to packs of 32 tablets in pharmacies, and 16 tablets in non-pharmacy outlets.[30] Up to 100 tablets may be sold in a single transaction. In Ireland, the limits are 24 and 12 tablets, respectively. In Australia, paracetamol tablets are available at supermarkets in small-pack sizes, whereas, with children's formulations, pack sizes greater than 48 tablets and suppositories are restricted to pharmacies.
Mechanism
Paracetamol is mostly converted to inactive compounds via Phase II metabolism by conjugation with sulfate and glucuronide, with a small portion being oxidized via the cytochrome P450 enzyme system. Cytochromes P450 2E1 (CYP2E1) and 3A4 (CYP3A4) convert paracetamol to a highly-reactive intermediary metabolite, N-acetyl-p-benzo-quinone imine (NAPQI).[31][32][33]
Under normal conditions, NAPQI is detoxified by conjugation with glutathione. In cases of paracetamol toxicity, the sulfate and glucuronide pathways become saturated, and more paracetamol is shunted to the cytochrome P450 system to produce NAPQI. As a result, hepatocellular supplies of glutathione become exhausted and NAPQI is free to react with cellular membrane molecules, resulting in widespread hepatocyte damage and death, leading to acute hepatic necrosis. In animal studies, hepatic glutathione must be depleted to less than 70% of normal levels before hepatotoxicity occurs.[31]
Toxic dose
The toxic dose of paracetamol is highly variable. In adults, single doses above 10 grams or 150 mg/kg have a reasonable likelihood of causing toxicity.[34] Toxicity can also occur when multiple smaller doses within 24 hours exceeds these levels, or even with chronic ingestion of doses as low as 4 g/day, and death with as little as 6 g/day. Consumption of alcohol has been tied to a smaller dose toxicity.
In children, acute doses above 200 mg/kg could potentially cause toxicity. This higher threshold is largely due to larger kidneys and livers relative to body size in children versus adults, and hence greater tolerance of paracetamol overdose than adults.[35] Acute paracetamol overdose in children rarely causes illness or death with chronic, supratherapeutic doses being the major cause of toxicity in children.
In a normal dose of 1 gram of acetaminophen four times a day, one-third of patients may have an increase in their liver function tests to three times the normal value.[36] However, it is unclear as to whether this leads to liver failure.[37]
Since paracetamol is often included in combination with other drugs, it is important to include all sources of paracetamol when checking a person's dose for toxicity. In addition to being sold by itself, paracetamol may be included in the formulations of various analgesics and cold/flu remedies as a way to increase the pain-relieving properties of the medication, and sometimes in combination with opioids such as hydrocodone to deter people from using it recreationally or becoming addicted to the opioid substance. In fact, the human toll of acetaminophen, in terms of both fatal overdoses and chronic liver toxicity to habitual abusers of pain medication, likely far exceeds the damage caused by the opioids themselves.[38
Risk factors
Chronic excessive alcohol consumption can induce CYP2E1, thus increasing the potential toxicity of paracetamol.[39] For this reason, analgesics such as aspirin or ibuprofen are often recommended over paracetamol for relief of hangovers when other factors, such as gastric irritation, are not involved.
Fasting is a risk factor, possibly because of depletion of hepatic glutathione reserves. It is well documented that concomitant use of the CYP2E1 inducer isoniazid increases the risk of hepatotoxicity, though whether 2E1 induction is related to the hepatotoxicity in this case is unclear.[40][41] Concomitant use of other drugs that induce CYP enzymes such as antiepileptics (including carbamazepine, phenytoin, and barbiturates) have also been reported as risk factors.
Natural history
Individuals that have overdosed on paracetamol, in general, have no specific symptoms for the first 24 hours. Although nausea, vomiting, and diaphoresis may occur initially, these symptoms, in general, resolve after several hours. After resolution of these symptoms, individuals tend to feel better, and may believe that the worst is over. If a toxic dose was absorbed, after this brief feeling of relative wellness, the individual develops overt hepatic failure. In massive overdoses, coma and metabolic acidosis may occur prior to hepatic failure.
In general, damage occurs in hepatocytes as they metabolize the paracetamol. Rarely, acute renal failure also may occur. This is usually caused by either hepatorenal syndrome or Multiple organ dysfunction syndrome. Acute renal failure may also be the primary clinical manifestation of toxicity. In these cases, it has been suggested that the toxic metabolite is produced more in the kidneys than in the liver.[42]
The prognosis of paracetamol toxicity varies depending on the dose and the appropriate treatment. In some cases, massive hepatic necrosis leads to fulminant hepatic failure with complications of bleeding, hypoglycemia, renal failure, hepatic encephalopathy, cerebral edema, sepsis, multiple organ failure, and death within days. In many cases, the hepatic necrosis may run its course, hepatic function may return, and the patient may survive with liver function returning to normal in a few weeks.
Diagnosis
Evidence of liver toxicity may develop in one to four days, although, in severe cases, it may be evident in 12 hours. Right-upper-quadrant tenderness may be present. Laboratory studies may show evidence of massive hepatic necrosis with elevated AST, ALT, bilirubin, and prolonged coagulation times (in particular, elevated prothrombin time). After paracetamol overdose, when AST and ALT exceed 1000 IU/L, paracetamol-induced hepatotoxicity can be diagnosed. However, the AST and ALT levels can exceed 10,000 IU/L. In general, the AST is somewhat higher than the ALT in paracetamol-induced hepatotoxicity.
A drug nomogram was developed in 1975, which estimated the risk of toxicity based on the serum concentration of paracetamol at a given number of hours after ingestion.[43] To determine the risk of potential hepatotoxicity, the paracetamol level is traced along the standard nomogram. A paracetamol level drawn in the first four hours after ingestion may underestimate the amount in the system because paracetamol may still be in the process of being absorbed from the gastrointestinal tract. Delay of the initial draw for the paracetamol level to account for this is not recommended, since the history in these cases is often poor and a toxic level at any time is a reason to give the antidote.
Treatment
Initial measures
The initial treatment for uncomplicated paracetamol overdose, similar to most other overdoses, is gastrointestinal decontamination. In addition, the antidote, acetylcysteine plays an important role. Paracetamol absorption from the gastrointestinal tract is complete within two hours under normal circumstances, so decontamination is most helpful if performed within this timeframe. Absorption may be somewhat slowed when it is ingested with food. There is considerable room for physician judgement regarding gastrointestinal decontamination; activated carbon administration is the most commonly-used procedure; however, gastric lavage may also be considered if the amount ingested is potentially life threatening and the procedure can be performed within 60 minutes of ingestion.[44] Syrup of ipecac has no role in paracetamol overdose because the vomiting it induces delays the effective administration of activated carbon and oral acetylcysteine.
Activated carbon adsorbs paracetamol, reducing its gastrointestinal absorption. Administering activated carbon also poses less risk of aspiration than gastric lavage. Previous to this method, there was reluctance to give activated carbon in paracetamol overdose, because of concern that it may also absorb acetylcysteine. Studies have shown that no more than 39% of an oral acetylcysteine is absorbed when they are administered together.[45] Other studies have shown that activated carbon seems to be beneficial to the clinical outcome. It appears that the most benefit from activated carbon is gained if it is given within two hours of ingestion.[46] However, administering activated carbon later than this can be considered in patients that may have delayed gastric emptying due to co-ingested drugs or following ingestion of sustained- or delayed-release paracetamol preparations. Activated carbon should also be administered if co-ingested drugs warrant decontamination. There are conflicting recommendations[45][47] regarding whether to change the dosing of oral acetylcysteine after the administration of activated carbon, and even whether the dosing of acetylcysteine needs to be altered at all.
Acetylcysteine
Acetylcysteine (also called N-Acetylcysteine or NAC) works to reduce paracetamol toxicity by supplying sulfhydryl groups (mainly in the form of glutathione, of which it is a precursor) to react with the toxic NAPQI metabolite so that it does not damage cells and can be safely excreted. (NAC can be bought as a dietary supplement in the United States.) If the patient presents less than eight hours after paracetamol overdose, then acetylcysteine significantly reduces the risk of serious hepatotoxicity. If NAC is started more than 8 hours after ingestion, there is a sharp decline in its effectiveness because the cascade of toxic events in the liver has already begun, and the risk of acute hepatic necrosis and death increases dramatically. Although acetylcysteine is most effective if given early, it still has beneficial effects if given as late as 48 hours after ingestion.[48] In clinical practice, if the patient presents more than eight hours after the paracetamol overdose, then activated carbon is probably not useful, and acetylcysteine is started immediately. In earlier presentations, the doctor can give carbon as soon as the patient arrives, start giving acetylcysteine, and wait for the paracetamol level from the laboratory.In United States practice, intravenous (IV) and oral administration are considered to be equally effective. However, IV is the only recommended route in Australasian and British practice.
Oral acetylcysteine is given as a 140mg/kg loading dose followed by 70 mg/kg every four hours for 17 more doses. Oral acetylcysteine may be poorly tolerated due to its unpleasant taste, odor, and its tendency to cause nausea and vomiting. It can be diluted to a 5% solution, from its marketed 10% or 20% solutions, to improve palatability. Where oral acetylcysteine is required, the inhalation formulation of acetylcysteine (Mucomyst) is often given orally.ol overdose increases two days after the ingestion, reaches a maximum on day four, and then gradually decreases. Patients with a poor prognosis are usually identified for likely liver transplantation. Acidemia is the most important single indicator of probable mortality and the need for transplantation. A mortality rate of 95% without transplant was reported in patients who had a documented pH less than 7.30. Other indicators of poor prognosis include renal insufficiency, grade 3 or worse hepatic encephalopathy, a markedly elevated prothrombin time, or a rise in prothrombin time from day three to day four. One study has shown that a factor V level less than 10% of normal indicated a poor prognosis (91% mortality), whereas a ratio of factor VIII to factor V of less than 30 indicated a good prognosis (100% survival).[50] The respiratory formulation can also be diluted and filter sterilized by a hospital pharmacist for IV use; however this is an uncommon practice. If repeat doses of carbon are indicated because of another ingested drug, then subsequent doses of carbon and acetylcysteine should be staggered every two hours.
Intravenous acetylcysteine (Parvolex/Acetadote) is used as a continuous intravenous infusion over 20 hours (total dose 300 mg/kg). Recommended administration involves infusion of a 150mg/kg loading dose over 15 minutes, followed by a 50mg/kg infusion over four hours; the last 100 mg/kg are infused over the remaining 16 hours of the protocol. Intravenous acetylcysteine has the advantage of shortening hospital stay, increasing both doctor and patient convenience, and it allows administration of activated carbon to reduce absorption of both the paracetamol and any co-ingested drugs without concerns about interference with oral acetylcysteine.[49]
Baseline laboratory studies include bilirubin, AST, ALT, and prothrombin time (with INR). Studies are repeated at least daily. Once it has been determined that a potentially-toxic overdose has occurred, acetylcysteine is continued for the entire regimen, even after the paracetamol level becomes undetectable in the blood. If hepatic failure develops, acetylcysteine should be continued beyond the standard doses until hepatic function improves or until the patient has a liver transplant.
Prognosis
The mortality rate from paracetamol overdose increases two days after the ingestion, reaches a maximum on day four, and then gradually decreases. Patients with a poor prognosis are usually identified for likely liver transplantation. Acidemia is the most important single indicator of probable mortality and the need for transplantation. A mortality rate of 95% without transplant was reported in patients who had a documented pH less than 7.30. Other indicators of poor prognosis include renal insufficiency, grade 3 or worse hepatic encephalopathy, a markedly elevated prothrombin time, or a rise in prothrombin time from day three to day four. One study has shown that a factor V level less than 10% of normal indicated a poor prognosis (91% mortality), whereas a ratio of factor VIII to factor V of less than 30 indicated a good prognosis (100% survival).[50]
Prevention
Besides preventing an overdose, one way to prevent liver damage may be the use of Paradote. Paradote is a combination tablet containing 100 mg methionine and 500 mg paracetamol (i.e., 20% methionine). Methionine is included in order to ensure that sufficient levels of glutathione in the liver are maintained in order to minimize the liver damage caused if a paracetamol overdose is taken.
Effects on animals
Paracetamol is extremely toxic to cats, and should not be given to them under any circumstances. Cats lack the necessary glucuronyl transferase enzymes to safely break paracetamol down and tiny fractions of a normal tablet for humans may prove fatal.[51] Initial symptoms include vomiting, salivation and discolouration of the tongue and gums. After around two days, liver damage is evident, typically giving rise to jaundice. Unlike an overdose in humans, it is rarely liver damage that is the cause of death, instead methaemoglobin formation and the production of Heinz bodies in red blood cells inhibit oxygen transport by the blood, causing asphyxiation. Effective treatment is occasionally possible for small doses, but must be extremely rapid.
In dogs, paracetamol is a useful anti-inflammatory with a good safety record, causing a lower incidence of gastric ulceration than NSAIDs. It should be administered only on veterinary advice. A paracetamol-codeine product (trade name Pardale-V)[52] licensed for use in dogs is available on veterinary prescription in the UK.[53]
Any cases of suspected ingestion in cats or overdose in dogs should be taken to a veterinarian immediately for detoxification.[54] The effects of toxicity can include liver damage, haemolytic anaemia, oxidative damage to the red blood cells and bleeding tendencies. There are no home remedies, and the amount of irreversible liver failure is dependent on how quickly veterinary intervention begins. Treatment of paracetamol overdose by a veterinarian may involve the use of supportive fluid therapy, acetylcysteine (trade name Mucomyst), methionine, or S-adenosyl-L-methionine (SAMe) to slow liver damage and cimetidine (trade name Tagamet) to protect against gastric ulceration. Once liver damage has occurred, it cannot be reversed.[55]
Paracetamol is lethal to snakes, also, and has been used in attempts to control the brown tree snake (Boiga irregularis) in Guam.[56]
REFERENCES
- Gormley, James J. "White willow bark is a gentle, effective pain-reliever." Better Nutrition. March, 1996.
- The Aspirin story. Did You Know?.
- Brodie BB, Axelrod J (1948). "The fate of acetanilide in man" (PDF). J Pharmacol Exp Ther 94 (1): 29-38.
- "A Festival of Analgesics." Chemical Heritage Foundation. 2001. Retrieved on August 17, 2007.
- (2002) Reader's Digest Guide to Drugs and Supplements. Pleasantville, New York; Montreal: Reader's Digest Association, Inc.. ISBN 0-7621-0366-3.
- Kis B, Snipes JA, Busija DW (2005). "Acetaminophen and the cyclooxygenase-3 puzzle: sorting out facts, fictions, and uncertainties". J. Pharmacol. Exp. Ther. 315 (1): 1-7. doi:10.1124/jpet.105.085431. PMID 15879007.
- Aronoff DM, Oates JA, Boutaud O (2006). "New insights into the mechanism of action of acetaminophen: Its clinical pharmacologic characteristics reflect its inhibition of the two prostaglandin H2 synthases". Clin. Pharmacol. Ther. 79 (1): 9–19. doi:10.1016/j.clpt.2005.09.009. PMID 16413237.
- Bertolini A, Ferrari A, Ottani A, Guerzoni S, Tacchi R, Leone S (2006). "Paracetamol: new vistas of an old drug". CNS drug reviews 12 (3-4): 250–75. doi:10.1111/j.1527-3458.2006.00250.x. PMID 17227290.
- Graham GG, Scott KF (2005). "Mechanism of action of paracetamol". American journal of therapeutics 12 (1): 46-55. PMID 15662292.
- Ohki S, Ogino N, Yamamoto S, Hayaishi O (1979). "Prostaglandin hydroperoxidase, an integral part of prostaglandin endoperoxide synthetase from bovine vesicular gland microsomes". J. Biol. Chem. 254 (3): 829–36. PMID 104998.
- Harvison PJ, Egan RW, Gale PH, Nelson SD (1986). "Acetaminophen as a cosubstrate and inhibitor of prostaglandin H synthase". Adv. Exp. Med. Biol. 197: 739–47. PMID 3094341.
- Roberts, L.J II. & Marrow, J.D. "Analgesic-antipyretic and Antiinflammatory Agents and Drugs Employed in the Treatment of Gout" in, "Goodman & Gilman's The Pharmacological Basis of Therapeutics 10th Edition" by Hardman, J.G. & Limbird, L.E. Published by McGraw Hill, 2001, p.687-731.
- , Jönsson BA, Ermund A, et al (2005). "Conversion of acetaminophen to the bioactive N-acylphenolamine AM404 via fatty acid amide hydrolase-dependent arachidonic acid conjugation in the nervous system". J. Biol. Chem. 280 (36): 31405–12. doi:10.1074/jbc.M501489200. PMID 15987694.
- Köfalvi A (2008). "Chapter 9: Alternative interacting sites and novel receptors for cannabinoid ligands. In: 'Cannabinoids and the Brain' Springer-Verlag": 131-160. doi:10.1007/978-0-387-74349_9.
- Ottani A, Leone S, Sandrini M, Ferrari A, Bertolini A (2006). "The analgesic activity of paracetamol is prevented by the blockade of cannabinoid CB1 receptors". Eur. J. Pharmacol. 531 (1-3): 280–1. doi:10.1016/j.ejphar.2005.12.015. PMID 16438952.
- Chandrasekharan NV, Dai H, Roos KL, et al (2002). "COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression". Proc. Natl. Acad. Sci. U.S.A. 99 (21): 13926–31. doi:10.1073/pnas.162468699. PMID 12242329.
- Borne, Ronald F. "Nonsteroidal Anti-inflammatory Drugs" in Principles of Medicinal Chemistry, Fourth Edition. Eds. Foye, William O.; Lemke, Thomas L.; Williams, David A. Published by Williams & Wilkins, 1995. p. 544-545.
- Dong H, Haining RL, Thummel KE, Rettie AE, Nelson SD (2000). "Involvement of human cytochrome P450 2D6 in the bioactivation of acetaminophen". Drug Metab Dispos 28 (12): 1397–400. PMID 11095574. Free full text
- Lee, William M. "Acetaminophen and the U.S. acute liver failure study group: Lowering the risks of hepatic failure." Hepatology. June 30, 2004. Vol. 40, Issue 1. p. 6-9.
- Bower, W.A.; Johns, M.; Margolis, H.S.; Williams, I.T.; Bell, B.P. "Population-Based Surveillance for Acute Liver Failure." Am. J. Gastroenterol.
- Mant, T.G.K.; Tempowski, J.H.; Volans, G.N.; Talbot, J.C.C. "Adverse reactions to acetylcysteine and effects of overdose." British Medical Journal. July 28, 1984. Vol. 289. pp. 217-219.
- Alsalim, W.; Fadel, M. "Oral methionine compared with intravenous n-acetyl cysteine for paracetamol overdose." Emerg. Med. J. 2003. Vol. 20. pp. 366-367.
- Bradley JD, Brandt KD, Katz BP, Kalasinski LA, Ryan SI (1991). "Comparison of an antiinflammatory dose of ibuprofen, an analgesic dose of ibuprofen, and acetaminophen in the treatment of patients with osteoarthritis of the knee". N. Engl. J. Med. 325 (2): 87-91. PMID 2052056.
- Clark E, Plint AC, Correll R, Gaboury I, Passi B (2007). "A randomized, controlled trial of acetaminophen, ibuprofen, and codeine for acute pain relief in children with musculoskeletal trauma". Pediatrics 119 (3): 460-7. doi:10.1542/peds.2006-1347. PMID 17332198.
- GarcÃa RodrÃguez LA, Hernández-DÃaz S (December 15, 2000). "The risk of upper gastrointestinal complications associated with nonsteroidal anti-inflammatory drugs, glucocorticoids, acetaminophen, and combinations of these agents". Arthritis Research and Therapy. PMID 11178116. Retrieved on 2006-12-29.
- Colás Chacartegui R, Temprano González R, Gómez Arruza C, Muñoz Cacho P, Pascual Gomez J (2005). "[Abuse pattern of analgesics in chronic daily headache: a study in the general population]". Rev Clin Esp 205 (12): 583-87. PMID 16527179.
- Sheen C, Dillon J, Bateman D, Simpson K, Macdonald T (2002). "Paracetamol toxicity: epidemiology, prevention and costs to the health-care system". QJM 95 (9): 609–19. PMID 12205339.
- Larson AM, Polson J, Fontana RJ, et al (2005). "Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study". Hepatology 42 (6): 1364-72. doi:10.1002/hep.20948. PMID 16317692.
- Ryder SD, Beckingham IJ (2001). "ABC of diseases of liver, pancreas, and biliary system. Other causes of parenchymal liver disease". BMJ 322 (7281): 290–92. doi:10.1136/bmj.322.7281.290. PMID 11157536. [11157536 Free full text]
- Limits on Sale in UK
- Richardson, J.A. Management of acetaminophen and ibuprofen toxicoses in dogs and cats. J Vet Emerg Crit Care. 2000; 10: 285–291.
- Rumbeiha, W.K.; Lin, Y.S.; Oehme, F.W. Comparison of N-acetylcysteine and methylene blue, alone or in combination, for treatment of acetaminophen toxicosis in cats. Am J Vet Res. 1995; 56: 1529–1533.
- Sellon, R.K. Acetaminophen. In: Peterson ME, Talcott PA, eds. Small Animal Toxicology. Toronto: WB Saunders, 2001: 388-395.
- Dart RC, Erdman AR, Olson KR, Christianson G, Manoguerra AS, Chyka PA, Caravati EM, Wax PM, Keyes DC, Woolf AD, Scharman EJ, Booze LL, Troutman WG; American Association of Poison Control Centers (2006). "Acetaminophen poisoning: an evidence-based consensus guideline for out-of- hospital management". Clin Toxicol (Phila) 44 (1): 1-18. PMID 16496488.
- Tenenbein M (2004). "Acetaminophen: the 150 mg/kg myth". J Toxicol Clin Toxicol 42 (2): 145-48. PMID 15214618.
- Watkins PB, Kaplowitz N, Slattery JT, et al (2006). "Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial". JAMA 296 (1): 87-93. doi:10.1001/jama.296.1.87. PMID 16820551.
- Dart RC, Bailey E (2007). "Does therapeutic use of acetaminophen cause acute liver failure?". Pharmacotherapy 27 (9): 1219-30. doi:10.1592/phco.27.9.1219. PMID 17723075.
- Brecher, Edward M (1972). Consumers Union Report on Licit and Illicit Drugs. Boston: Little, Brown.
- Zimmerman HJ, Maddrey WC (1995). "Acetaminophen (paracetamol) hepatotoxicity with regular intake of alcohol: analysis of instances of therapeutic misadventure". Hepatology 22 (3): 767-73. PMID 7657281.
- Crippin JS (1993). "Acetaminophen hepatotoxicity: potentiation by isoniazid". Am J Gastroenterol 88 (4): 590-92. PMID 8470644.
- Nolan CM, Sandblom RE, Thummel KE, Slattery JT, Nelson SD (1994). "Hepatotoxicity associated with acetaminophen usage in patients receiving multiple drug therapy for tuberculosis". Chest 105 (2): 408–11. PMID 7508362.
- Boutis K, Shannon M (2001). "Nephrotoxicity after acute severe acetaminophen poisoning in adolescents". J Toxicol Clin Toxicol 39 (5): 441-“5. PMID 11545233.
- Rumack B, Matthew H (1975). "Acetaminophen poisoning and toxicity". Pediatrics 55 (6): 871–76. PMID 1134886.
- Vale JA, Kulig K; American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists (2004). "Position paper: gastric lavage". J Toxicol Clin Toxicol 42 (7): 933-43. PMID 15641639.
- Ekins B, Ford D, Thompson M, Bridges R, Rollins D, Jenkins R (1987). "The effect of activated charcoal on N-acetylcysteine absorption in normal subjects". Am J Emerg Med 5 (6): 483-87. PMID 3663288.
- Buckley NA, Whyte IM, O'Connell DL, Dawson AH. (1999). "Activated charcoal reduces the need for N-acetylcysteine treatment after acetaminophen (paracetamol) overdose". J Toxicol Clin Toxicol 37 (6): 753-57. PMID 10584587.
- Spiller H, Krenzelok E, Grande G, Safir E, Diamond J (1994). "A prospective evaluation of the effect of activated charcoal before oral N-acetylcysteine in acetaminophen overdose". Ann Emerg Med 23 (3): 519-23. PMID 8135427.
- Keays R, Harrison P, Wendon J, Forbes A, Gove C, Alexander G, Williams R (1991). "Intravenous acetylcysteine in paracetamol induced fulminant hepatic failure: a prospective controlled trial". BMJ 303 (6809): 1026–9. PMID 1954453.
- Buckley N, Whyte I, O'Connell D, Dawson A (1999). "Oral or intravenous N-acetylcysteine: which is the treatment of choice for acetaminophen (paracetamol) poisoning?". J Toxicol Clin Toxicol 37 (6): 759-67. PMID 10584588.
- Pereira LM, Langley PG, Hayllar KM, Tredger JM, Williams R (1992). "Coagulation factor V and VIII/V ratio as predictors of outcome in paracetamol induced fulminant hepatic failure: relation to other prognostic indicators". Gut 33 (1): 98-102. PMID 1740285. Full text at PMC: 1373872
- Allen AL (2003). "The diagnosis of acetaminophen toxicosis in a cat". Can Vet J 44 (6): 509-10. PMID 12839249.
- Pardale-V Tablets: Presentation. UK National Office of Animal Health Compendium of Animal Medicines (September 28, 2006).
- Pardale-V Tablets: Legal Category. UK National Office of Animal Health Compendium of Animal Medicines (November 15, 2005).
- Villar D, Buck WB, Gonzalez JM (1998). "Ibuprofen, aspirin and acetaminophen toxicosis and treatment in dogs and cats". Vet Hum Toxicol 40 (3): 156-62. PMID 9610496.
- Manning AM. Acetaminophen Toxicity in Dogs. PetPlace.com.
- Johnston J, Savarie P, Primus T, Eisemann J, Hurley J, Kohler D (2002). "Risk assessment of an acetaminophen baiting program for chemical control of brown tree snakes on Guam: evaluation of baits, snake residues, and potential primary and secondary hazards". Environ Sci Technol 36 (17): 3827-“33. PMID 12322757.