B.1.1.529 Omicron Variant
SEE LATEST COVID UPDATES -- VARIANTS, VACCINES TREATMENT AND WORLD NEWS
NEW PUBLICATION --Dec. 21, 2021 --PreRelease SARS-CoV-2 Omicron Variant: ACE2 Binding, Cryo-EM Structure of Spike Protein- ACE2 Complex and Antibody Evasion
---"DATA AVAILABILITY" PDB Files... Cryo-EM reconstructions and atomic models generated during this study are available at the PDB and EMBD databases under the following accession codes: Unbound Omicron spike protein trimer (PDB ID 7T9J, EMD- EMDB 25759), global ACE2-bound Omicron spike protein trimer ((PDB ID 7T9K, EMD-25760), focus-refinement of the ACE2-RBD interface for the ACE2-bound Omicron spike protein trimer (PDB ID 7T9L, EMD-25761)...."
The new variant appears to have over 30 spike mutations. South African scientists are worried and several of them have warned that the new variant has a combination of mutations that can possibly cause the virus to evade immunity.
What are the mutations in B.1.1.529 that are of conern?
The mutation P681H seen in the new variant has also been reported in Alpha, Mu, some Gamma, and B.1.1.318 variants. The new variant also carries the N679K, N501Y, P681H, N679K, D614Gvariants. Studies have shown that this mutation helps the variant be more transmissible. It also allows the virus to readily bind to the human angiotensin-converting enzyme 2 (ACE2) receptors.
Note: May take 5-10 seconds to load Jsmol File below
PDB:7v7f-->mutated RBD to Omicron B.1.1.529- RBD(15 amino acids)= G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H
-------------->spin on -------->- spin off->zoom in to E484A -------->- zoom out
To Rotate the Molecule--->Left Click and Drag
To Zoom-->>Left Click + hold Shift button and Drag Vertically
To see amino acid and atom number hold cursor over atom
Jmol Menu --->>Right-Click
Variant-- World Health Organization
Only four variants of the coronavirus prior to Omicron were designated as variants of concern namely Alpha (lineage B.1.1.7, the so-called 'UK variant'), Beta (lineage B.1.351, the so-called 'South Africa variant'), Gamma (lineage P.1, the so-called 'Brazil variant') and Delta (lineage B.1.617.2).The Omicron variant has a large number of mutations, of which some are concerning. Many of those mutations had not been observed in other strains. The variant is characterised by 30 amino acid changes, three small deletions and one small insertion in the spike protein compared with the original novel virus, of which 15 are located in the receptor binding domain (residues 319-541). It also carries a number of changes and deletions in other genomic regions. Additionally, the variant has three mutations at the furin cleavage site. The furin cleavage site increases SARS-CoV-2 infectivity. The mutations by genomic region are the following:
Spike protein: A67V, Δ69-70, T95I, G142D, Δ143-145, Δ211, L212I, ins214EPE, G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H, T547K, D614G, H655Y, N679K, P681H, N764K (furin cleavage site), D796Y, N856K, Q954H, N969K, L981F Half (15) of these 30 changes are located in the receptor binding domain-RBD (residues 319-541)
RBD = G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H,
FURIN CLEAVAGE SITE MUTATIONS --Variant contains not one, but two furin cleavage site mutations—P681H & N679K—this is the first time @PeacockFlu [Imperial virologist Tom Peacock] seen 2 of these mutations in a single variant. Furin site spells trouble," Feigl-Ding said.
The 3 amnino acid insertion (ins214EPE) in Omicron
Omicron variant of SARS-CoV-2 harbors a unique insertion mutation of putative viral or human genomic origin
Is this why virus is more transmissable but less severe?
Abstract -- Pre Print
"...The emergence of a heavily mutated SARS-CoV-2 variant (B.1.1.529, Omicron) and it'sspread to 6 continents within a week of initial discovery has set off a global public health alarm. Characterizing the mutational profile of Omicron is necessary to interpret its shared or distinctive clinical phenotypes with other SARS-CoV-2 variants. We compared the mutations of Omicron with prior variants of concern (Alpha, Beta, Gamma, Delta), variants of interest (Lambda, Mu, Eta, Iota and Kappa), and all 1523 SARS-CoV-2 lineages constituting 5.4 million SARS-CoV-2 genomes. Omicron's Spike protein has 26 amino acid mutations (23 substitutions, two deletions and one insertion) that are distinct compared to other variants of concern. Whereas the substitution and deletion mutations have appeared in previous SARS-CoV-2 lineages, the insertion mutation (ins214EPE) has not been previously observed in any SARS-CoV-2 lineage other than Omicron. The nucleotide sequence encoding for ins214EPE could have been acquired by template switching involving the genomes of other viruses that infect the same host cells as SARS-CoV-2 or the human transcriptome of host cells infected with SARS-CoV-2. For instance, given recent clinical reports of co-infections in COVID-19 patients with seasonal coronaviruses (e.g. HCoV-229E), single cell RNA-sequencing data showing co-expression of the SARS-CoV-2 and HCoV-229E entry receptors (ACE2 and ANPEP) in respiratory and gastrointestinal cells, and HCoV genomes harboring sequences homologous to the nucleotide sequence that encodes ins214EPE, it is plausible that the Omicron insertion could have evolved in a co-infected individual. There is a need to understand the function of the Omicron insertion and whether human host cells are being exploited by SARS-CoV-2 as an 'evolutionary sandbox' for host-virus and inter-viral genomic interplay..." see full paper --excellent read
Comparison to Delta Variant
"...Pre-activated virus To penetrate cells, the SARS-CoV-2 spike protein must be cut twice by host proteins. In the SARS-CoV-1 virus that causes severe acute respiratory syndrome (SARS), both incisions occur after the virus has locked on to a cell. But with SARS-CoV-2, the presence of the furin cleavage site means that host enzymes (including one called furin) can make the first cut as newly formed viral particles emerge from an infected cell. These pre-activated viral particles can then go on to infect cells more efficiently than do particles requiring two cuts, says Whittaker...."
"...Shi’s team and other groups have zeroed in on a mutation that alters a single amino acid in the SARS-CoV-2 spike protein — the viral molecule responsible for recognizing and invading cells. The change, which is called P681R and transforms a proline residue into an arginine, falls within an intensely studied region of the spike protein called the furin cleavage site..."
Follow-up experiments by both groups showed that the P681R change was largely responsible for spike being clipped so much more efficiently.
Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant
---->>>"...The Delta spike has accumulated mutation P681R located at a furin cleavage site that separates the spike 1 (S1) and S2 subunits. Reverting the P681R mutation to wild-type P681 decreases effects----->WILL P681H, N764K (furin cleavage site) FOR OMICRON COMPETE WITH THE P681 CLEAVAGE OF P681 IN DELTA....we need some good modeling studies here.
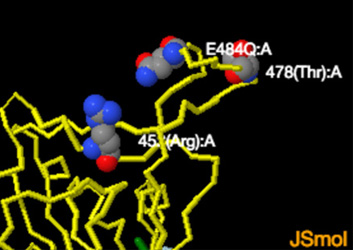
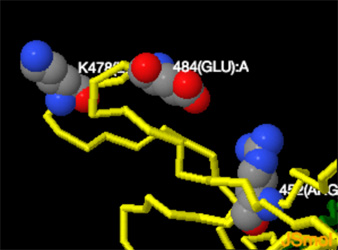
How Dangerous is the Delta Variant (B.1.617.2)
The receptor binding domain is the portion of the spike protein that binds directly to human ACE2 receptors. Delta has 3 RBD mutations. The first, a lysine to asparagine substitution at position 417, is present in some, but not all sequences of B.1.617.2. It is also common to the Beta variant and has been associated with conformational changes to S protein, which may aid in immune escape. The second mutation, a leucine to arginine substitution at position 452, is common to the former variant of interest Epsilon, and is known to increase affinity for ACE2 receptors found on the surface of a variety of human cells, including the lungs. And the third, a threonine to lysine substitution at position 478, is common to the B.1.1.519 lineage, and has been predicted to increase electrostatic potential and steric hindrance, which may further increase RBD/ACE2 binding affinity and enable immune escape. ..."How Dangerous Is the Delta Variant (B.1.617.2)?
-------------->spin on -------->- spin off
-------------->zoom into receptor binding site
-(Amino acids 417K ,478T, 452L)
------->- zoom out
Jmol Menu --->>Right-Click
-------------->spin on -------->- spin off
-------------->zoom into receptor binding site
-(Amino acids 417N (this is delta+), 478K, 452R ---Delta with K417N originally corresponded to lineages AY.1 and AY.2
------->- zoom out
Jmol Menu --->>Right-Click
Readings:
Where did 'wierd' Omicron come from?(Science Journal) December 2, 2021
Heavily mutated Omicron variant puts scientists on alert November 2021
The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets April 2021
Evolutionary analysis of the Delta and Delta Plus variants of the SARS-CoV-2 viruses Nov. 2021
SARS-CoV-2 Variant Classifications and Definitions Oct. 2021
Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant --Oct 2021
Structural and functional mechanism of SARS-CoV-2 cell entry
Effectiveness of Covid-19 Vaccines against the B.1.617.2 (Delta) Variant Aug. 2021
Sars-CoV-2 Variants
The novel coronavirus SARS-CoV2
B.1.1.7 and B.1.525 UK variant
B.1.427/B.1.429, California QP77P Mutation
B.1.526, NY E484K or S477N Mutations
Molecules of Disease
-
Small Molecules
Cholesterol
Nicotine
Trans Fatty Acid
Alcohol
Acetyaldehyde
- Proteins
Cytokines
Hemoglobin S
Prions
Botulinum Toxin
Explain it with Molecules
- Why is water such a good solvent?
- Why does ice float?
- Why do solids, liquids and gases behave differently?
- What is the geometry of methane?
- What's the difference between alpha and beta glucose?
- How does caffeine work in the brain?
- How does soap work?
- What is the difference between sucrose and fructose?
- Why is carbon monoxide so dangerous?
- Why is graphite so soft if it is made of only carbon?
- What is the difference between Carbyne and Graphite?
- Why is the fullerene and similar structures the cornerstone of nanotechnology?
- How big is a nanotube?
- Why does table salt have a cubic crystal shape?
- What is the structure of the benzene molecule?
- Why do carcinogens cause cancer?
- What causes Sickle Cell Anemia?
- What is the difference between sodium nitrite and nitrate?
- How do drugs work?