Novel coronavirus SARS-CoV2
The Spike Protein
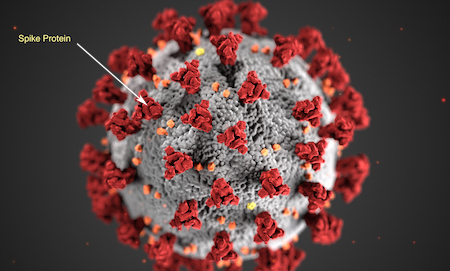
Sars-CoV-2 virus shows the spike proteins -- trimer visible at arrow end (original Image from fda.gov)
There are thousands of different coronaviruses currently in existence. Four of them are responsible for the common cold. Recently several more dangerous viruses have emerged, namely SARS (Fuhan, China) which killed more than 700 people and MERS (Saudi Arabia). Although MERS was the most deadly with 35% of patients dying, it was difficult to pass on from person to person.
The current coronavirus pandemic started in Wuhan, China is far deadlier than previous coronavirus in part because it is able to lie undetected in a host yet be transmissible. In addition after the virus enters the body it binds to its target ACE2 (angiotensin-converting enzyme 2) which are not just found in the respiratory system but throughout the body. Binding of the spike protein to its receptor human ACE2 is through its receptor-binding domain (RBD),which is a 273 amino acid segment of the whole spike protein stretching from residue 319 to 591 (see fig.1 below)
Binding of the spike protein to its receptor human ACE2 is through its receptor-binding domain (RBD),which is a 273 amino acid segment of the whole spike protein stretching from residue 319 to 591 shown in red CPK (spacefill) for the B Chain PDB:6Zgg
To Rotate the Molecule--->Left Click and Drag
To Zoom-->>Left Click + hold Shift button and Drag Vertically
Jmol Menu --->>Right-Click
The spike (S) protein of SARS-CoV-2 is significant since it mediates receptor binding and cell entry and is the , dominant target of the immune system. It exhibits substantial conformational flexibility, transitioning from a closed to open conformations to expose its receptor-binding site.
The RBD of the homotrimeric SARS-CoV-2 sages hetepike protein switches between 'up' and 'down' conformations like a hinge. ACE2 can only interact with the RBD when it is in the 'up' conformation.
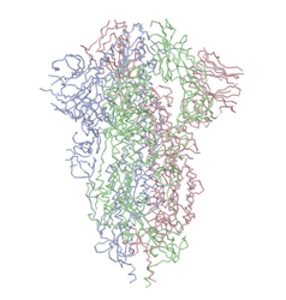
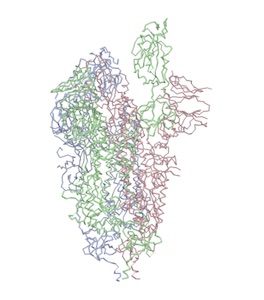
Above: left image: closed state pdb:6vxx; right image open state pdb:6vyb)
"...The virus binds to host cells through its trimeric spike glycoprotein, making this protein a key target for potential therapies and diagnostics. Wrapp et al. determined a 3.5-angstrom-resolution structure of the 2019-nCoV trimeric spike protein by cryo–electron microscopy. Using biophysical assays, the authors show that this protein binds at least 10 times more tightly than the corresponding spike protein of severe acute respiratory syndrome (SARS)–CoV to their common host cell receptor..." (Wrapp et al., 2020)
"...The total length of SARS-CoV-2 S is 1273 amino acids and consists of a signal peptide (amino acids 1–13) located at the N-terminus, the S1 subunit (14–685 residues), and the S2 subunit (686–1273 residues); the last two regions are responsible for receptor binding and membrane fusion, respectively...".(See Review article by Huang et al., Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19
Figure 1.
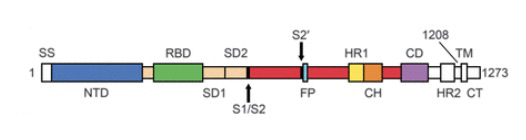
Above image: shows amino acids labeled showing NTD (N-Terminal Domain), RBD (Reaction Binding Domain) located in S1 domain and components of S2 domain. For a full explanation see: Wrapp et al., (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation.
Pre and Post Fusion Spike Structures
"...The trimeric viral spike (S) protein catalyzes fusion between viral and target cell membranes to initiate infection. Here, we report two cryo–electron microscopy structures derived from a preparation of the full-length S protein, representing its prefusion (2.9-angstrom resolution) and postfusion (3.0-angstrom resolution) conformations, respectively. The spontaneous transition to the postfusion state is independent of target cells. The prefusion trimer has three receptor-binding domains clamped down by a segment adjacent to the fusion peptide. The postfusion structure is strategically decorated by N-linked glycans, suggesting possible protective roles against host immune responses and harsh external conditions. These findings advance our understanding of SARS-CoV-2 entry and may guide the development of vaccines and therapeutics..." source: Distinct conformational states of SARS-CoV-2 spike protein
The atomic structure coordinates have been deposited in the RCSB Protein Data Bank (PDB) under the accession numbers 6XR8 and 6XRA, --(Lab of Bing Chen )
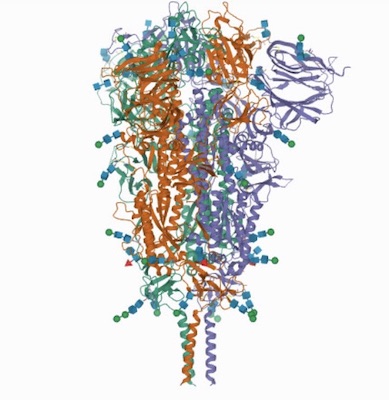
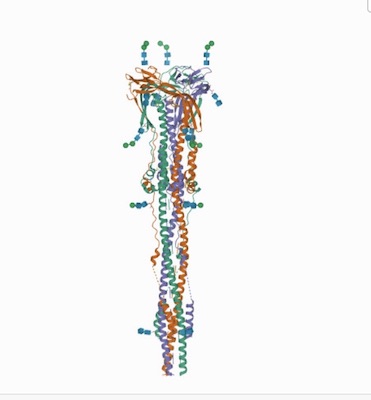
Above: Pre and post fusion --Distinct conformational states of SARS-CoV-2 spike protein. Cai, Y., Zhang, J., Xiao, T., Peng, H., Sterling, S.M., Walsh Jr., R.M., Rawson, S., Rits-Volloch, S., Chen, B. (2020) Science 369: 1586-1592.
Stabilizing the Spike Protein in Prefusion Conformation
To stabilize prefusion spike proteins a 2-Proline addition was added at amino acid numbers 986 and 987 in many pdb structures. This same kink technique was used for stabilizing the mRNA vaccines for Pfizer and Moderna.
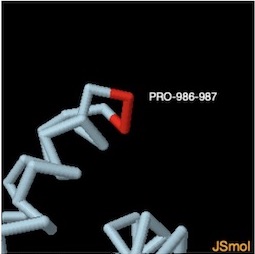
PROLINE 986-987 ON CHAIN A
Use ZOOM IN button on Jsmol file at left to view in 3D.
Mutations and Escape Mutations
SARS-CoV-2 has been mutating since it was first sequenced in early January 2020. Since SARS-CoV-2 has a genetic proofreading mechanism achieved by non-structure protein (NSP) 14 in synergy with NSP10 and NSP12 it has a higher fidelity in its transcription and replication process than that of other single-stranded RNA viruses. Nonetheless, 7123 single mutations have been detected in the 12,754 US SARS-CoV-2 strains in the past few months with respect to the first SARS-CoV-2 genome collected on 24 December 2019. Source: Analysis of SARS-CoV-2 mutations in the United States suggests presence of four substrains and novel variants.
Escape Mutations
In addition SARS-CoV-2 tends to delete portions of RNA that encode the N-terminal domain (NTD) of the spike, a mutation that its proofreader can't detect. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape.
Some of the most concerning new variants, including B.1.1.7, initially identified in the UK, and B.1.351, first spotted in South Africa, have one or two of these short deletions in the NTD, along with a cluster of mutations in the receptor binding domain (RBD) of the spike, notably the E484K, which must attach to the ACE2 receptor in order to enter host cells.
The E484K is called an escape mutation because it also helps the virus escape attack from antibodies. With this mutation, the virus can slip past our immune defenses and make us sick. Escape mutations arise as pressure is put on the virus to survive.
Sars-CoV-2 Variants
The novel coronavirus SARS-CoV2
B.1.1.7 and B.1.525 UK variant
B.1.427/B.1.429, California QP77P Mutation
B.1.526, NY E484K or S477N Mutations
Molecules of Disease
-
Small Molecules
Cholesterol
Nicotine
Trans Fatty Acid
Alcohol
Acetyaldehyde
- Proteins
Cytokines
Hemoglobin S
Prions
Botulinum Toxin
Explain it with Molecules
- Why is water such a good solvent?
- Why does ice float?
- Why do solids, liquids and gases behave differently?
- What is the geometry of methane?
- What's the difference between alpha and beta glucose?
- How does caffeine work in the brain?
- How does soap work?
- What is the difference between sucrose and fructose?
- Why is carbon monoxide so dangerous?
- Why is graphite so soft if it is made of only carbon?
- What is the difference between Carbyne and Graphite?
- Why is the fullerene and similar structures the cornerstone of nanotechnology?
- How big is a nanotube?
- Why does table salt have a cubic crystal shape?
- What is the structure of the benzene molecule?
- Why do carcinogens cause cancer?
- What causes Sickle Cell Anemia?
- What is the difference between sodium nitrite and nitrate?
- How do drugs work?